The Food and Drug Administration (FDA) plays a critical role in safeguarding public health by regulating the pharmaceutical industry. This article examines the FDA’s evolution, its current functions in drug development and approval, and its initiatives to modernize regulatory processes using information technology. We will explore how the FDA balances rigorous safety standards with the need for timely access to effective treatments, with a focus on What Is The Purpose Of Food And Drug Administration.
The FDA’s Role in Public Health
With a significant portion of the US population relying on prescription medications, the FDA’s role in ensuring drug safety and efficacy is paramount. The FDA strives to ensure that drugs on the US market meet stringent standards. This review aims to provide insights into the journey of a chemical compound from laboratory inception to FDA-approved therapeutic drug.
Organizational Structure of the FDA
As a division of the US Department of Health and Human Services, the FDA comprises five directorates, including the Office of Medical Products and Tobacco. The Center for Drug Evaluation and Research (CDER) is a key subsection responsible for regulating over-the-counter and prescription drugs.
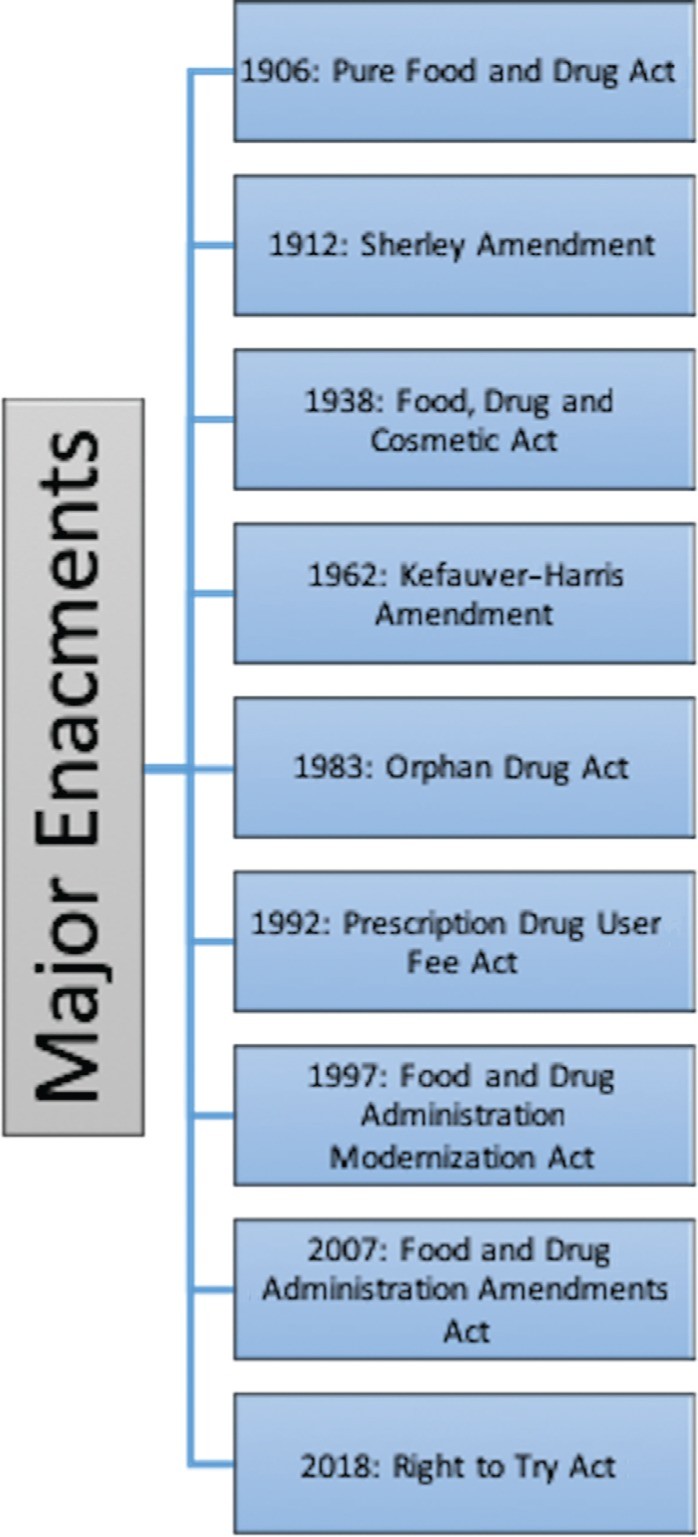
Historical Milestones in Drug Regulation
Early 20th-century drug products often featured false labels, unregulated ingredients, and unsubstantiated therapeutic claims. The 1906 Pure Food and Drug Act marked a turning point, prohibiting the interstate sale of misbranded food and drugs.
The Pure Food and Drug Act
The Sherley Amendment of 1912 further outlawed false therapeutic claims on drug labels. These laws were enforced by the Bureau of Chemistry, which later became the FDA in 1930.
The Food, Drug, and Cosmetic Act (FD&C Act)
The 1938 FD&C Act was enacted following a tragic incident in 1937, where 105 people died from taking elixir sulfanilamide. This led to requirements for establishing drug safety before market release.
The Kefauver-Harris Amendment
In 1962, the Kefauver-Harris Amendment mandated that drug manufacturers prove both safety and efficacy before marketing a drug. This amendment also required reporting of adverse effects and obtaining informed consent from clinical trial participants, setting the stage for modern randomized controlled trials.
Subsequent Legislation
The 1983 Orphan Drug Act incentivized the development of drugs for rare diseases. The 1992 Prescription Drug User Fee Act allowed the CDER to collect fees from drug companies, expediting the approval process. Enhanced postmarketing surveillance was introduced with the Adverse Event Reporting System (AERS) in 1998, and the ClinicalTrials.gov website was launched in 2000 to improve public access to clinical trial information. The FDA Amendments Act of 2007 further expanded postmarketing safety activities through the REMS program. The Right to Try Act, enacted in 2018, allows physicians to seek expanded access to drugs that have completed phase I trials, bypassing FDA approval in certain cases.
The Drug Development and Approval Process
The drug development process begins with identifying a chemical compound with therapeutic potential through extensive laboratory testing. Promising compounds then undergo preclinical in vivo testing in animals to assess safety and determine a safe starting dose for human trials.
Investigational New Drug (IND) Application
The FDA’s involvement begins with the submission of an IND application. This application includes preclinical data, manufacturing information, and proposed clinical study protocols.
Clinical Trial Phases
Phase I trials focus on determining safe dosages and collecting pharmacokinetic data in healthy volunteers. Phase II trials evaluate drug efficacy and side effects in patients with the targeted disease. Phase III trials, the most extensive, are often double-blinded, randomized, and controlled, involving thousands of patients to assess efficacy across different populations and identify less common adverse events.
New Drug Application (NDA) and Approval
Communication between the drug sponsor and the FDA is ongoing, with formal meetings before and after phase III trials. The NDA is the formal request for drug approval, including all trial data. The FDA reviews the NDA to determine if the drug’s benefits outweigh its risks. If approved, the drug can be marketed in the US.
Marketing Approval in the US and Europe
In the US, FDA approval allows drug companies to negotiate pricing with insurers and government programs. In Europe, the European Medicines Agency (EMA) evaluates drugs, and the European Commission authorizes market approval. However, additional cost-effectiveness analyses, such as those conducted by the UK’s National Institute for Health and Care Excellence (NICE), may be required before a drug is available to the public.
Expedited Approval Pathways
Orphan Drug Act
The Orphan Drug Act provides incentives for developing drugs targeting rare diseases, where traditional market incentives are lacking.
Fast-Track, Accelerated Approval, and Priority Review
These pathways expedite the approval of drugs for serious or life-threatening conditions that meet unmet medical needs. Accelerated approval relies on surrogate endpoints, requiring postmarket phase V trials to confirm clinical benefits. Priority review aims to complete NDA reviews within six months.
Breakthrough Therapy
The “breakthrough therapy” designation, part of the FDA Safety and Innovation Act of 2012, facilitates the fastest development times for drugs that show substantial improvement over current therapies.
Expanded Access Programs
The FDA’s expanded-access program allows patients with serious conditions to apply for access to experimental therapies before FDA approval.
Cardiac Safety and the Critical Path Initiative
The FDA introduced the Critical Path Initiative (CPI) in 2004 to modernize the drug development process. The Cardiac Safety Research Consortium (CSRC) is a public-private partnership focused on researching the cardiac safety of medical drugs and devices.
Proarrhythmic Potential and TQT Studies
The evaluation of a drug’s proarrhythmic potential involves thorough QT/QTc (TQT) studies, which assess the drug’s effect on the QT/QTc interval on an ECG. Preclinical testing includes in vitro electrophysiology studies to assess the ability of a substance to inhibit the hERG current.
Comprehensive In Vitro Proarrhythmia Assay (CiPA)
CiPA is a new initiative that aims to more directly test mechanisms responsible for torsades, offering a more precise assessment of proarrhythmia risk.
Conclusion
The Food and Drug Administration balances rigorous standards for drug efficacy and safety with the need for timely access to effective treatments. Through partnerships and research, the FDA continually seeks to improve the cost-effectiveness and efficiency of the drug development process. The question “what is the purpose of food and drug administration” boils down to ensuring public safety while fostering innovation in the pharmaceutical industry.